Journal of Medical Sciences and Health
DOI: 10.46347/jmsh.v10.i3.24.109
Year: 2024, Volume: 10, Issue: 3, Pages: 339-343
Case Report
Paheli Maru1 , Dhaval Jetly2 , Rujuta Shah3 , Sanjiban Patra1 , Udaya S4
1Assistant Professor (DM Oncopathology), Oncopathology Department, Gujarat Cancer Research Institute, Ahmedabad, Gujarat, India,
2Professor, Oncopathology Department, Gujarat Cancer Research Institute, Ahmedabad, Gujarat, India,
3Assistant Professor, Oncopathology Department, Gujarat Cancer Research Institute, Ahmedabad, Gujarat, India,
4Senior Resident, DM Oncopathology, Oncopathology Department, Gujarat Cancer Research Institute, Ahmedabad, Gujarat, India
Address for correspondence:
Dhaval Jetly, Professor, Oncopathology Department, Gujarat Cancer Research Institute, Ahmedabad, Gujarat, India.
E-mail: [email protected]
Received Date:07 July 2024, Accepted Date:02 September 2024, Published Date:18 November 2024
Paediatric pancreatic tumours are rare and mostly malignant. Because they present with vague clinical features, it is important to recognise them and make correct pathological diagnoses using radiological suggestions and histopathology. A one-year-old male child presented to the surgical outpatient department with complaints of abdominal pain for 4 months. The ultrasonography showed a lesion in the left suprarenal region. An ultrasound-guided trucut biopsy showed tumour cells with predominant glandular (acinar) and focal solid (squamoid) patterns with areas of necrosis and haemorrhage. A diagnosis of epithelial cell tumour was given, and IHC was advised. The PET CT scan showed an avidly heterogeneously enhanced large, well-defined soft tissue density lesion involving the left upper abdomen, measuring 5.6x5.6x4.9 cm. The lesion showed an internal zone of necrosis, haemorrhage, and calcification. However, no radiological possibility of a malignant mass lesion of the pancreas was suggested. On immunohistochemistry, the tumour cells were positive for AE1, CK7, beta-catenin, and AFP, along with Ki67, in 70% of tumour nuclei. Based on the morphology, immunohistochemistry, and clinicopathological findings, a diagnosis of pancreatoblastoma was given, and serum AFP levels were suggested. The mass was completely excised, and typical morphological features of the tumor were identified. The definite diagnosis may be challenging, especially when dealing with limited tumour tissue in biopsies. Integration of clinical, radiological, and histopathological findings is essential in pancreatoblastoma diagnosis. In cases of unusual presentation with vague radiologic findings, thorough knowledge of the histopathological features along with a precise IHC panel may help arrive at the diagnosis.
Keywords: Pancreatoblastoma, Clinicoradiology, Histopathology, IHC, Survival
Pancreatic tumours in children are infrequently encountered in clinical practice and are grouped according to the cell of origin. 1 Those arising from the exocrine epithelial cells include acinar cell adenocarcinoma, neuroendocrine carcinoma, solid pseudopapillary neoplasm, ductal adenocarcinoma, islet cell tumour, and pancreatoblastoma. Among these, pancreatoblastoma is the most common malignant epithelial neoplasm 2, 3 .
We present the case of a 1-year-old male child who presented to the surgical outpatient department with complaints of abdominal pain due to a gradually enlarging mass for 4 months. The ultrasonography showed a lesion in the left suprarenal region (the adrenal gland was not seen separately from the lesion) with internal foci of calcification. The lesion abutted the spleen, left kidney, and inferior surface of the liver. An ultrasound-guided trucut biopsy was performed, and we received multiple grey-white linear cores measuring 0.5 to 1.0 cm long with a
The patient underwent a PET CT scan before the biopsy, which showed FDG avidly heterogeneously enhancing a large, well-defined soft tissue density lesion involving the left upper abdomen and measuring 5.6x5.6x4.9 cm. The lesion showed an internal zone of necrosis, haemorrhage, and calcification and abutted the stomach, bowel loops, spleen, body, and tail of the pancreas. The adrenal gland was seen separately from the lesion. However, no radiological possibility of a malignant mass lesion of the pancreas was suggested.
On histopathological examination, the tumour cells showed predominant glandular (acinar) and focal solid (squamoid) patterns with areas of necrosis and haemorrhage. A diagnosis of epithelial cell tumour was given, and IHC was advised which included AE1, CK7, EMA, CEA, synaptophysin, chromogranin, beta-catenin, and AFP, considering the differential diagnosis of pancreatoblastoma, acinar cell adenocarcinoma and neuroendocrine carcinoma.
On IHC, the tumour cells were positive for AE1, CK7, beta-catenin, and AFP, along with Ki67, in 70% of tumour nuclei. Based on the morphology, IHC, and clinicopathological findings, a diagnosis of pancreatoblastoma was given, and serum AFP levels were suggested, which were only mildly increased to 91.1 ng/mL.
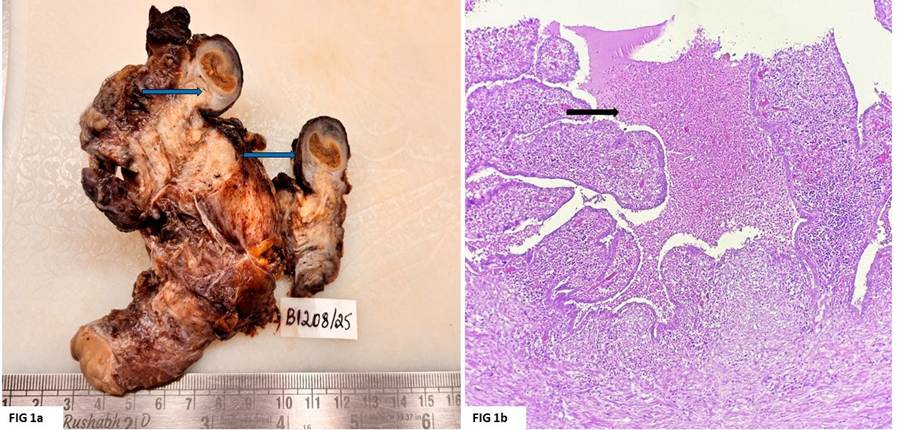
This was followed by the excision of the pancreatic mass and peripancreatic nodes. The mass measured 5.5x5x3.2cm. On cut section, the tumour was solid, grey-white, well-circumscribed, lobulated with a focal gritty area (1.2 cm) and traversed by thin fibrous septa. On microscopic examination of multiple sections, a circumscribed capsulated tumour was seen compressing the pancreatic parenchyma. The tumour consisted of hypercellular epithelial lobules separated by fibrous bands. The cells were arranged in acinar and trabecular patterns and mixed with nests of squamoid cells. The tumour cells were monomorphic polygonal cells with abundant eosinophilic cytoplasm, vesicular chromatin, and conspicuous nucleoli. Focal neuroendocrine differentiation was also seen. Areas of calcification were noted. Necrosis was evident. Mitosis amounted to 8–9/10 hpf. Lymphovascular invasion was evident. but the perineural invasion was not seen. All submitted peripancreatic nodes were free of tumours (0–10).


Immunohistochemistry was performed on a Ventana Benchmark XT auto immunostainer. The clones used for various immunohistochemical markers are as follows: synaptophysin (SP11), chromogranin (LK2H10), Wilm Tumour 1 (6F-H2), AE1 (AE1/AE3), CK7 (OV-TL12/30), beta-catenin (4), EMA (29), CEA (II-7), and AFP (polyclonal).
Primary pancreatic tumours in children are rare, with an overall age-adjusted incidence of 0.018 new cases per 100,000 paediatric patients. Their non-specific clinical presentation and overlapping imaging characteristics often make an accurate preoperative diagnosis difficult. Also, the prognosis varies according to the histologic type, so an accurate and early diagnosis may aid in the appropriate management and survival of these patients.
The most common morphological pattern- the acinar cell pattern is seen in acinar cell carcinoma, neuroendocrine tumors and pancreatoblastoma. Acinar cell carcinoma shows high cellularity, tumour cells in a solid, cord-like or acinar-like arrangement with little stroma, and monotonous tumour cells with a single distinct nucleolus. They are positive for markers like trypsin, chymotrypsin, lipase, BCL10 and PASD. Neuroendocrine tumours resemble small-cell-type neuroendocrine carcinoma and are positive for markers like synaptophysin, chromogranin, INSM1 and AFP, if serum AFP levels are elevated. Pancreatoblastoma comprises epithelial tumour cells arranged in a trabecular pattern with squamous nests. Apart from the acinar and neuroendocrine markers, positivity for CK5, EMA and beta catenin is seen in squamous nests.
Surgery of the first case of pancreatoblastoma was done by Becker 4 in 1957 in an infant. However, the first histomorphological description was given by Frabel et al. 5 in 1971, and only about 200 cases have been reported since then, more commonly in Asians. 6
Pancreatoblastoma is more frequent in males, with the median age of presentation being 5 years. It is a slow-growing tumour with typical presentation as a large palpable abdominal mass and abdominal pain, with or without vomiting. Tumour rupture and haemorrhage have also been reported. Previous studies reported elevated serum AFP levels in most cases. 7 In this case, AFP levels were suggested to support our biopsy interpretation.It has been associated with Beckwith-Wiedemann and Familial Adenomatous Polyposis syndromes. 8
The aim of radiology in patients presenting with an abdominal mass is to determine the site of origin of the tumour and to establish the extent of the disease, including the presence or absence of metastases. Pancreatoblastomas are large, well-defined, at least partially circumscribed, heterogeneous masses with low to intermediate signal intensity on T1-W images and high signal intensity on T2-W images. Contrast medium enhancement is common on CT and MR images. 9 Liver, lung, lymph nodes, spleen, and bone have been the reported sites of metastases in pancreatoblastoma. 10 But in our case, the radiology did not localise the tumour to the pancreas, and there was no evidence of metastases.
A definitive diagnosis of pancreatoblastoma is usually made after histopathological examination of laparotomy or resection specimens because it relies on the identification of epithelial differentiation, including acinar, glandular, and trabecular architectures, as well as squamoid differentiation, so-called squamoid corpuscles. However, in our case, despite the unusual presentation, the diagnosis was made on biopsy.
Recently there have been reports of adult pancreatoblastoma with subtle symptoms,lack of characteristic clinical manifestations and atypical histological morphology combined with familial adenomatous polyposis.11, 12 We thus want to highlight that one must be aware of the histomorphological features of this rare tumour as it may help in making an early diagnosis of a large retroperitoneal mass with vague radiological findings.
The mainstay of treatment is complete surgical resection, either upfront or following neoadjuvant chemotherapy. When the tumour is encapsulated, a complete removal is possible, and thus the curability rate is high. However, these neoplasms display more aggressive behaviour and an overall poorer prognosis in adults than in children, probably because they are diagnosed in the later stages of the disease. The most commonly used chemotherapy regimens are doxorubicin and cisplatin. 7 Our case had a well-encapsulated tumour and did not require neoadjuvant chemotherapy before resection. The parents were advised for long-term follow-up to treat any early local recurrence or metastasis.
Pancreatoblastoma tends to be diagnosed at an advanced stage when the tumour has extended beyond the pancreas or is metastatic. It should be considered even in small biopsies with a glandular and solid pattern when physical examination shows an asymptomatic mass in the upper abdomen with an elevated serum AFP level and no definite radiologic evidence. Early diagnosis when the tumour is still encapsulated may aid in complete resection and the patient's survival.
IHC: immunohistochemistry; AE1: anticytokertan monoclonal antibody; CK7: cytokeratin; EMA: epithelial membrane antigen; CEA: carcinoembryonic antigen; AFP: alpha-fetoprotein; PET-CT: positron emission tomography and computed tomography; FDG: fluorodeoxyglucose.
Paheli Maru and Udaya S: acquisition of the data.
Paheli Maru: analysis and research of the literature, and preparation of the manuscript.
Dhaval Jetly: conception and design of the manuscript and critical revision of the manuscript for intellectual content.
Rujuta Shah and Sanjiban Patra: critical revision of the manuscript for intellectual content.
Dhaval Jetly: administrative support and supervision. All authors read and approved the final manuscript.
The authors received no financial support for the research, authorship, and/or publication of this manuscript.
The study has been approved by the institute research committee of GCRI with reference number IRC/2023/P-38, ensuring compliance with legal and ethical criteria in the study.
The authors declare that they have no competing interests.
Subscribe now for latest articles and news.