Journal of Medical Sciences and Health
DOI: 10.46347/jmsh.2020.v06i01.007
Year: 2020, Volume: 6, Issue: 1, Pages: 30-32
Case Report
Rashmi Sriram1, H Harshavardhan Gowda2, Rajendra Okade3, Mouryabha Shale4, Kranthi Varma5
1Assistant Professor, Department of Dermatology, BGS Global Institute of Medical Sciences, Bengaluru, Karnataka, India,
2Senior Resident, Department of Dermatology, BGS Global Institute of Medical Sciences, Bengaluru, Karnataka, India,
3Professor & Head, Department of Dermatology, BGS Global Institute of Medical Sciences, Bengaluru, Karnataka, India,
4Assistant Professor,Department of Dermatology, Adichunchanagiri Institute of Medical Sciences, Mandya, Karnataka, India,
5Junior resident,Department of Dermatology, Navodaya Medical College, Raichur, Karnataka, India
Address for correspondence:
H Harshavardhan Gowda, Department of Dermatology, BGS Global Institute of Medical Sciences, Bengaluru, Karnataka, India. Phone: +91-9743815454. E-mail: [email protected]
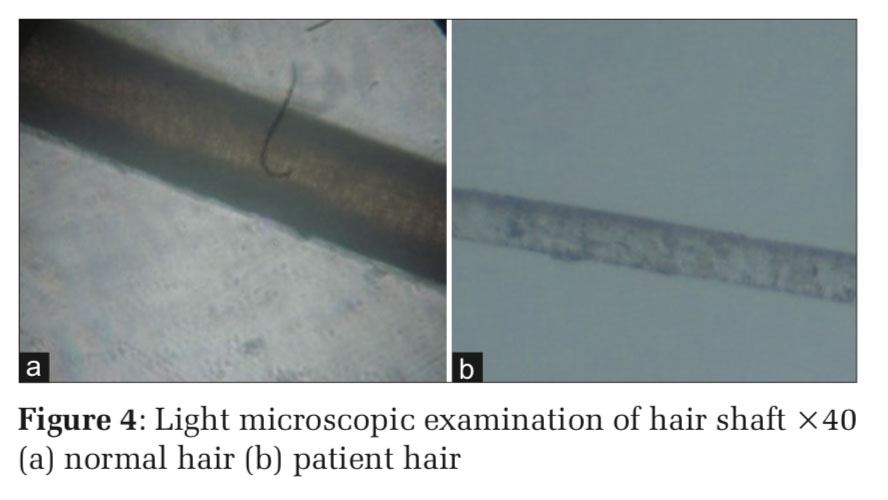
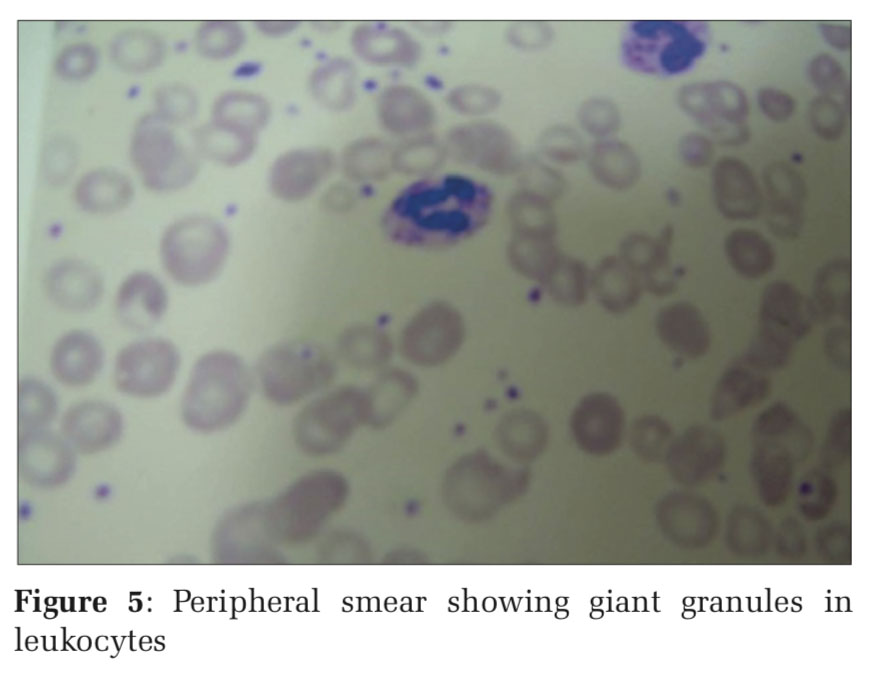
A 4-year-old female child born out of a consanguineous marriage, full-term normal vaginal delivery, presented with multiple hypopigmented macules giving mottled pigmentation over the face and neck. There was a history of swelling and pain in the right ear and recurrent skin infections. On examination, she had a silvery gray hair, mottled pigmentation over the face and neck, perichondritis of right ear, impetiginous lesions over the lower limbs, hepatomegaly. Ophthalmological examination revealed partial oculocutaneous albinism and nystagmus. Routine blood investigations were within normal limits. Peripheral smear examination revealed prominent granules within the leukocytes. Hair shaft examination revealed regularly arranged small clumps of melanin. It was diagnosed as a case of Chediak–Higashi syndrome. This case is presented for its rarity.
KEY WORDS: Chediak–Higashi, silvery hair, syndrome.
Chediak–Higashi syndrome (CHS) is a rare lysosomal disorder characterized by defect is in the gene lysosome trafficking regulator, resulting in defective vesicular transport to and from the lysosome. Chediak–Higashi gene product has been identified and mapped on chromosome 1 q 43.[1] It is characterized by oculocutaneous hypopigmentation, photophobia, nystagmus, neutropenia, and an abnormal susceptibility to cutaneous and respiratory infections.[2,3] Hepatosplenomegaly, lymphadenopathy, pancytopenia, jaundice, and gingivitis with bleeding tendency are other common features.[4] The diagnosis can be confirmed by recognition of the characteristic large cytoplasmic inclusions in leukocytes by peripheral smear examination.[1] Morbidity results from patients to frequent bacterial infections or to an accelerated phase, consisting of a lymphoproliferative syndrome with hemophagocytosis and infiltration of most tissues.[3]
A 4 year old female child born at full term by normal delivery to consanguineous parents presented with a history of pigmentary disturbances in skin from the age of 2 years. There was a history of recurrent skin infections, swelling, and pain in the right ear for 15days. No history of respiratory infection, bleeding, and similar complaints in the family. On examination, she had silvery gray hair, multiple ill- defined hypopigmented macules over the face, neck, perichondritis of right ear, multiple erosions covered with crusts over both upper limbs and lower limbs, hepatomegaly (Figures 1 and 2). Blood investigations were within normal limits. Hair shaft examination revealed regularly arranged clumps of melanin (Figures 3 and 4). Peripheral smear showed giant granules in leukocytes (Figure 5). It was diagnosed as a case of CHS. Appropriate antibiotics were used to treat the infection. The patient was referred to a higher center for further management.
Patients with CHS exhibit hypopigmentation of the skin, hair, and eyes due to the presence of giant melanosomes which cause pigment dilution, possibly secondary to impaired melanin trans-port.[5] There are recurrent staphylococcal and streptococcal infections as a result of neutropenia and other complications such as anemia, thrombocytopenia, and lymphoma can also occur in these patients.[3,4] In our case, the child had silvery gray hair, recurrent skin infections, hepatomegaly, perichondritis of the right ear, and typical peripheral smear examination showing giant granules within the leukocytes and hair shaft light microscopy with findings consistent with a diagnosis of CHS. The condition has to be differentiated from other conditions such as Griscelli syndrome and Elejalde disease. Certain laboratory investigations help to differentiate between the three entities [Table 1].
The prognosis of CHS is generally not good.[2-4] Clinicians should be familiar with the severity of the disease because death often occurs in the first decade of life as a result of overwhelming infections, hemorrhage, or development of the accelerated lymphoma-like phase.[2-4] Treatment is limited to allogeneic bone marrow transplantation (BMT), although the accelerated phase may respond to etoposide plus systemic steroids and intrathecal methotrexate, the disease relapses invariably.[3]
Allogeneic BMT from a human leukocyte antigen- matched sibling is the therapy of choice and should perform early. If no matched family donor is available, an unrelated donor or a placental blood graft is a good alternative. Without BMT, children with Chediak–Higashi syndrome usually die before age 10 years.[7]




Subscribe now for latest articles and news.