Journal of Medical Sciences and Health
DOI: 10.46347/jmsh.v12.i1.25.285
Year: 2026, Volume: 12, Issue: 1, Pages: 99-103
Short communications/brief reports
R Sindhu 1, Champaka Gopal 2, Akkamahadevi S Patil 2, Suma Mysore Narayana 3, Ashwini Nargund 3, Geeta V Patil Okaly 3
1Consultant Oncopathologist, GANGA Hospitals, Coimbatore, Tamil Nadu, India.
2Professor, Department of OncoPathology, Kidwai Memorial Institute of Oncology, Bengaluru, Karnataka, India.
3Associate Professor, MD, Department of OncoPathology, Kidwai Memorial Institute of Oncology, Bengaluru, Karnataka, India.
Address for correspondence: R Sindhu, Consultant Oncopathologist, GANGA Hospitals, Coimbatore, Tamil Nadu, India.
E-mail: [email protected]
Received Date:13 August 2025, Accepted Date:06 October 2025, Published Date:15 May 2026
Anaplastic large cell lymphoma (ALCL) is an aggressive subtype of non-Hodgkin lymphoma predominantly involving lymph nodes and skin. Extranodal or extracutaneous presentations are rare and diagnostically challenging due to their varied histomorphology. We present a retrospective series of 16 cases of extranodal ALCL involving diverse anatomical sites including bone, soft tissue, scalp, sinonasal region, retroperitoneum, endobronchial region, trachea, and breast. The patient cohort had a 1:1 male-to-female ratio, with equal representation of pediatric and adult cases. Diagnosis relied on biopsy and immunohistochemistry (IHC), with ALK and CD30 positivity as hallmarks. Histomorphological variants included common, lymphohistiocytic, small cell, and composite patterns. Treatment consisted of age-appropriate chemotherapy with or without radiotherapy. Ten patients achieved remission, four died of which one had progressive disease and two were lost to follow-up. This study underscores the pleomorphic nature of extranodal ALCL and highlights the importance of timely diagnosis and comprehensive IHC workup for effective management.
Anaplastic large cell lymphoma (ALCL) accounts for 2–8% of adult and approximately 30% of childhood non-Hodgkin lymphomas.[1] It was first characterized in 1985 by its expression of CD30 (Ki-1 antigen) and cohesive proliferation of large pleomorphic cells.[2] The WHO classification recognizes four ALCL subtypes: primary systemic ALK-positive (ALK+), primary systemic ALK-negative, primary cutaneous ALCL, and breast implant-associated ALCL.[3] Extranodal presentations are poorly documented, mainly appearing in isolated case reports. Diagnosing ALCL in unusual locations is particularly challenging due to overlapping histological features with sarcomas, melanomas, and poorly differentiated carcinomas. Prompt diagnosis is critical as ALCL responds well to chemotherapy and radiotherapy, potentially avoiding unnecessary surgeries.
This retrospective study was conducted at our institution over five and a half years, from January 2017 to June 2022. It included 16 histologically confirmed cases of extranodal/extracutaneous ALCL from departmental archives. Patient demographics, clinical presentation, histological patterns, IHC profiles, treatment regimens, and follow-up outcomes were collected and analyzed. Informed consent from patients was waived, as this was a retrospective study.
A total of 16 histologically confirmed cases of extranodal ALCL were included in this study. The male-to-female ratio was 1:1, and the age of the patients ranged from 11 to 74 years, with a mean age of 24 years. The cohort was equally divided between pediatric (<18 years) and adult (>18 years) cases, with eight patients in each group. The most commonly involved site was soft tissue (7 cases), followed by bone and scalp (2 cases each). Single cases were reported in the sinonasal region, retroperitoneum, endobronchial region, trachea, and breast.
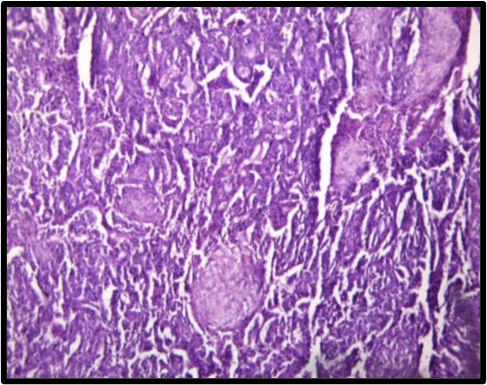
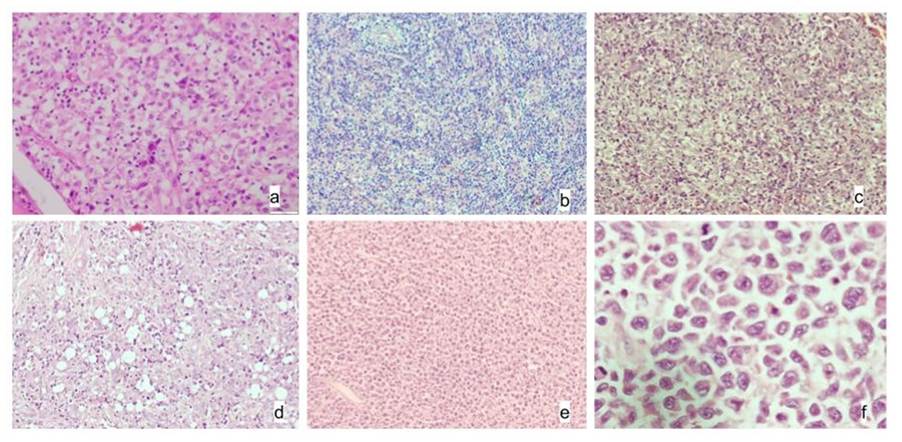
Histopathological examination revealed that the common pattern was predominant in 9 cases, characterized by diffuse sheets of monomorphic round cells. Among these, four cases exhibited classic hallmark cells, while the remaining five showed varying degrees of pleomorphism, featuring multilobated or horseshoe-shaped nuclei, wreath-like nuclear configurations.
| Cas No. |
Age/ Gender |
Site of Presentation |
Morpholoical Differentials |
Histological Pattern |
Treatment | Follow Up |
|---|---|---|---|---|---|---|
| 1. | 14/M | Scalp | Neuroblastoma Embyonal rhabdomyosarcoma | Common- monomorphic cells | ALCL 99 -PROTOCOL | Remission |
| 2. | 17/M | Shoulder |
Lymphoma Histiocytic neoplasm |
Lymphohistiocytic | ALCL 99 -PROTOCOL | Died after 1 year of diagnosis |
| 3. | 30/F | Retroperitoneum | Lymphoma | Small cell | CHEMO | Remission |
| 4. | 74/F | Leg | Soft tissue sarcoma | Common- monomorphic cells | LEFT TREATMENT | Denied treatment- death in 2 months |
| 5. | 16/F | Thigh | Soft tissue sarcoma, Metastasis | Composite- common and lymphohistiocytic | SURGERY+ 5#CHOP | Remission |
| 6. | 29/M | Multiple bone lytic lesion | Metastasis | Common- monomorphic cells | - | Lost follow-up |
| 7. | 14/F | Endobronchial lesion | Neuroendocrine tumor, lymphoma | Small cell | 2#CT- DEATH | Died within 2 months of diagnosis |
| 8. | 26/F | Breast | Lymphoma, Poorly differentiated carcinoma | Common | 6# CHOP+E- | Remission |
| 9. | 17/M | Sinonasal mass | SRCT/papilloma/sinonasal undifferentiated carcinoma | Common | 3#CHOP+IT MTX +RT | Remission |
| 10. | 18/M | D12 vertebra-bone | SRCT/metastasis | Common | CHEMO | Remission |
| 11. | 38/M | Left thigh | Sarcoma/Lymphoma | Composite- common and lymphohistiocytic | SURGERY+ 5#CHOP | Remission |
| 12. | 11/F | Scalp |
Neuroblastoma/ metastasis/ lymphoma |
Small cell | ALCL 99 protocol | Remission |
| 13. | 13/F | Thigh | Metastatic carcinoma/Lymphoma | Common- monomorphic cells | ALCL 99 PROTOCOL | Remission |
| 14. | 23/F | Thigh | Sarcoma/histiocytic lesion | Lymphohistiocytic |
5#CT CHOP+ 1#BRENTUXIMAB |
Died due to progression of disease |
| 15. | 20/M | Bilateral gluteal mass | High grade sarcoma with epithelioid morphology | Common | ON TREATMENT | Remission |
| 16. | 25/M | Posterior wall of trachea | Poorly differentiated neoplasm | Common | - | Lost follow up |
"doughnut" cells, and embryoid forms with convoluted nuclei and having abundant eosinophilic cytoplasm.
The small cell variant was identified in 3 cases, presenting as small round blue cells closely resembling other small round cell tumors or lymphomas. The lymphohistiocytic pattern was seen in 2 cases, composed of sheets and clusters of histiocytes interspersed with small lymphoid cells, morphologically simulating a histiocytic neoplasm. Additionally, 2 cases displayed a composite pattern, combining features of both the common and lymphohistiocytic subtypes. The initial morphological differential diagnoses considered include Ewing sarcoma, neuroblastoma, rhabdomyosarcoma, small round cell tumors, metastatic carcinoma, lymphoma and histiocytic neoplasms [Table. 1], [Fig. 1].
| Case No. | LCA | CD3 | CD4 | CD8 | ALK* | EMA | CD30 | Ki 67% | Other Positive Markers |
|---|---|---|---|---|---|---|---|---|---|
| 1 | + | + | N+C | D | + | 60% | CD99 | ||
| 2 | + | + | N+C | ND | + | ND | Granzyme | ||
| 3 | + | + | + | N+C | D | + | 40% | Granzyme, perforin, | |
| 4 | F | - | N+C | D | + | ND | Granzyme-focal | ||
| 5 | + | + | ++ | + | N+C | F | + | 80% | CD7, CD99 |
| 6 | + | + | N+C | D | + | 65% | Bcl 6 | ||
| 7 | + | - | N+C | D | + | 70% | - | ||
| 8 | + | + | + | ++ | Membrane | D | + | 90% | CD5, Bcl6, Bcl2, MUM1 |
| 9 | + | + | Cytoplasmic | D | + | 80% | CD5, CD56, CD43 | ||
| 10 | + | + | + | N+C | F | + | 80% | - | |
| 11 | + | + | N+C | ND | + | 55% | - | ||
| 12 | + | ND | N+C | D | + | 90% | - | ||
| 13 | + | + | N+C | ND | + | 90% | - | ||
| 14 | F | + | Cytoplasmic | - | + | 90% | - | ||
| 15 | + | + | N+C | F | + | 90% | - | ||
| 16 | + | - | N+C | ND | + | 85% | MUM1 |
*- Pattern of ALK expression. N- nuclear, C- cytoplasmic, F- focal, D- diffuse, ND- not done
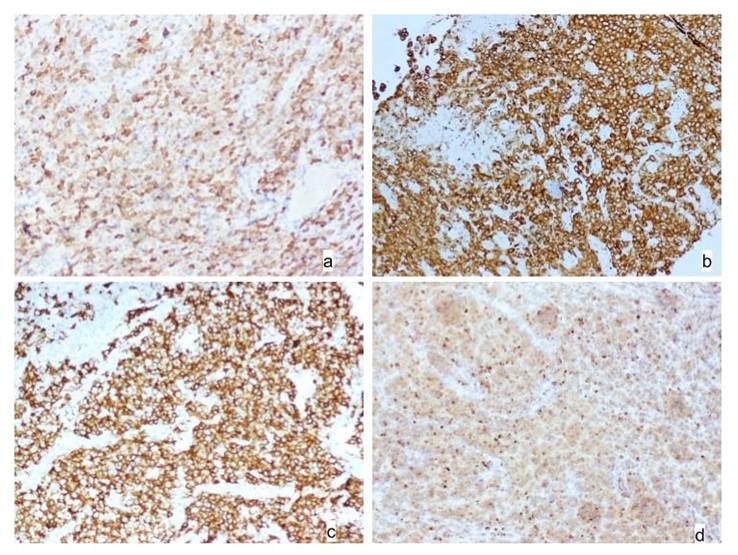
Immunohistochemical analysis showed uniform CD30 positivity across all cases. ALK expression was observed in 13 cases with both nuclear and cytoplasmic staining, 2 cases with cytoplasmic-only expression, and 1 case exhibited a membranous pattern [Fig. 2]. EMA was positive in 11 of 16 cases. Interestingly, 3 cases displayed a null phenotype, being negative for CD3. The Ki-67 proliferation index ranged from 40% to 90%, indicating high mitotic activity. Additionally, some cases showed
aberrant expression of markers such as CD99, MUM1, Bcl-6, CD56, CD43, and CD138 [Table. 2].
With regard to disease spread, case 6 exhibited bone marrow involvement, while case 14 had sternal, mediastinal and axillary lymph node involvement and brain metastases with progression of disease. Three cases (Case 13,14 and 15) demonstrated infradiaphragmatic lymph node involvement on CT imaging, further indicating systemic spread.
Treatment regimens were age-specific. Pediatric patients were managed with the ALCL-99 protocol, while adults received CHOP-E chemotherapy with or without adjunctive radiotherapy. Of the 16 patients, ten achieved complete remission. Four patients succumbed to the disease, of which three showed lymphohistiocytic and one had small cell pattern—highlighting their poorer prognosis. One patient showed progression of the disease despite treatment, and two patients were lost to follow-up.
Extranodal ALCL presents a unique diagnostic challenge due to its ability to mimic other malignancies both clinically and histologically. Our cohort of 16 cases of extranodal/extracutaneous ALCL spans a wide demographic and anatomical range, reflecting the disease’s heterogeneity. Our study showed equal pediatric and adult distribution of extranodal cases. However, in general higher incidence was noted in children and young adults, as well as its strong ALK association in pediatric cases.[3] In our series, all cases were ALK-positive, reinforcing the correlation between extranodal presentation and ALK positivity.
Extranodal ALCL often masquerades as non-lymphoid malignancies. Gustafson et al. described pediatric soft-tissue ALCL misdiagnosed as neuroblastoma or rhabdomyosarcoma—mirroring our findings where initial differentials included sarcomas, neuroblastoma, and small round cell tumors.[4] This emphasizes the imperative of considering ALCL in the differential diagnosis of soft-tissue tumors, especially in children. The morphologic mimicry observed in our cohort underscores the role of hallmark cells (horseshoe/wreath-like nuclei) and CD30 positivity with or without ALK positivity as essential diagnostic clues. In our series, extranodal involvement included rare and diagnostically challenging sites such as the endobronchial region, trachea, retroperitoneum, and breast. These findings are consistent with sporadic case reports that highlight ALCL's potential to involve virtually any anatomic location, sometimes without overt lymphadenopathy.[5, 6] In our series, two cases presented as primary osseous lesions. Similar rare reports by Gaulard et al. and Swerdlow et al. describe ALCL masquerading as primary bone tumors, closely resembling sarcomas, Ewing family tumors, or metastatic carcinomas necessitating detailed immunohistochemical workup.[7, 8] One notable case involved an endobronchial lesion in a young male, initially suspected to be a neuroendocrine tumor. Literature describing primary pulmonary ALCL is sparse, with isolated reports like those by Kanagal-Shamanna et al. underlines the importance of CD30 and ALK expression in such contexts.[5] Likewise, our breast ALCL case, although rare, aligns with the literature on breast implant-associated ALCL but differed due to the absence of implant history, echoing findings by Laurent et al. of de novo ALCL in mammary tissue.[6] The sinonasal and retroperitoneal locations also posed diagnostic challenges. Prior studies, including those by Abenoza et al. and Campo et al., have stressed that sinonasal ALCL may mimic undifferentiated carcinomas or extranodal NK/T-cell lymphomas.[9, 10] Our case demonstrated high Ki-67 and CD30/ALK positivity, aiding in confirmation.
Histologically, the small cell and lymphohistiocytic variants in our cohort showed aggressive behavior, corroborating with existing literature suggesting poorer prognosis in these subtypes.[11] These patterns often obscure the characteristic hallmark cells of ALCL and may show CD30 expression in scattered areas unlike the diffuse expression in other patterns, increasing the risk of misdiagnosis. The high Ki-67 proliferation index (40–90%) reflects aggressive disease, consistent with reports of advanced-stage presentation in extranodal ALCL.[12] Extranodal presentations—bone marrow (case 6), CNS metastases (case 14), and infradiaphragmatic lymphadenopathy—highlight the disease’s capacity for systemic spread, in line with reports showing up to 40% bone marrow involvement and frequent extranodal engagement in organs such as bone, soft tissue, lung, and liver.[13] Three of our cases demonstrated a null phenotype (CD3-negative), a feature that further complicates lineage determination. This phenotype, although recognized in ALCL, often necessitates exclusion of histiocytic lesions and poorly differentiated carcinomas. This is further complicated by EMA positivity that is observed in ALCL. Hence, broad panel of markers including CD68, CD163, PanCK, CD30, other T cell markers and cytotoxic cell markers are required often to make an accurate diagnosis.[8] Therapeutically, age-appropriate regimens (ALCL-99 in children, CHOP-E in adults ± radiotherapy) were used. Remission in 10 patients (62.5%) aligns with literature on ALK-positive ALCL, which has superior outcomes compared to ALK-negative cases.[14, 15] Notably, the effective implementation of age-tailored chemotherapy protocols resulted in high remission rates, underscoring the curability of ALCL when diagnosed accurately and early.
While treatment outcomes were not the primary focus of our study, the observed remission and mortality patterns provide contextual support for the prognostic implications of histologic patterns. Morphological diversity mandates a high index of suspicion, while ALK positivity heralds a more favorable prognosis.
Extranodal ALCL is a rare but clinically significant variant of non-Hodgkin lymphoma. This study highlights the broad histological spectrum and diverse clinical presentations, particularly in uncommon anatomical sites. Accurate and timely diagnosis using a comprehensive IHC approach is crucial for guiding effective therapy. Awareness of its morphological and immunophenotypic variability can prevent misdiagnosis and improve patient outcomes.
1. Gaidano G, Dalla-Favera R. Pathobiology of non-Hodgkin lymphomas. In: Hoffman R, Benz EJ Jr, Shattil SJ, Furie B, Cohen HJ, Silberstein LE, editors. Hematology: Basic Principles and Practice. 3rd ed. New York: Churchill Livingstone; 2000. p. 1213-29.
2. The expression of the Hodgkin's disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood. 1985; 66 (4). Available from: https://doi.org/10.1182/blood.v66.4.848.848
3. Campo E, Jaffe ES, Cook JR, Quintanilla-Martinez L, Swerdlow SH, Anderson JR, et al. WHO classification of tumours: Haematolymphoid tumours. 5th ed. Vol. 1. Lyon: International Agency for Research on Cancer; 2022. p. 473-91
4. Anaplastic large cell lymphoma: another entity in the differential diagnosis of small round blue cell tumors. Annals of Diagnostic Pathology. 2009; 13 (6). Available from: https://doi.org/10.1016/j.anndiagpath.2009.09.002
5. Kanagal-Shamanna R, Port JD, Brynes RK, Medeiros LJ. Anaplastic large cell lymphoma presenting as an endobronchial mass. Ann Diagn Pathol. 2009;13(5):313-7
6. Breast implant-associated anaplastic large cell lymphoma: Two distinct clinicopathological variants with different outcomes. Annals of Oncology. 2015; 27 (2). Available from: https://doi.org/10.1093/annonc/mdv575
7. Expression of T-cell-associated antigens by anaplastic large-cell lymphomas of the B-cell phenotype: A clinicopathologic and immunophenotypic study of 40 cases. American Journal of Surgical Pathology. 1998; 22 (12). Available from: https://doi.org/10.1172/jci115044
8. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Revised 4th ed. Lyon: IARC Press; 2017
9. Abenoza P, Vardiman JW. Anaplastic large cell lymphoma mimicking undifferentiated carcinoma of the sinonasal tract. Cancer. 1993;72(3):882-7.
10. Campo E, Gaulard P, Pileri SA, Stein H, Jaffe ES, Müller-Hermelink HK, et al. Aggressive T-cell lymphomas (excluding ALCL): Advances in biology and clinical management. Blood. 1999;93(11):3681-95
11. Prognostic significance of histologic subtypes in ALK-positive anaplastic large-cell lymphoma: A study of 235 cases. Blood. 1999; 93 (11). Available from: https://doi.org/10.1182/blood.v93.11.3913.411k22_3913_3921
12. Clinical and laboratory characteristics of systemic anaplastic large cell lymphoma in Chinese patients. Journal of Hematology & Oncology. 2012; 5 (1). Available from: https://doi.org/10.1186/1756-8722-5-38
13. The Pathological Spectrum of Systemic Anaplastic Large Cell Lymphoma (ALCL). Cancers. 2018; 10 (4). Available from: https://doi.org/10.3390/cancers10040107
14. Prognostic Impact of Morphologic and Phenotypic Features of Childhood ALK-Positive Anaplastic Large-Cell Lymphoma: Results of the ALCL99 Study. Journal of Clinical Oncology. 2011; 29 (35). Available from: https://doi.org/10.1200/jco.2011.36.5411
15. ALK-positive anaplastic large-cell lymphoma in adults: an individual patient data pooled analysis of 263 patients. Haematologica. 2019; 104 (12). Available from: https://doi.org/10.3324/haematol.2018.213512
Subscribe now for latest articles and news.