Journal of Medical Sciences and Health
DOI: 10.46347/jmsh.2016.v02i03.008
Year: 2016, Volume: 2, Issue: 3, Pages: 36-39
Case Report
Kunal Sharma1 , S R Negi2 , Rajni Joshee2 , Kanchan Rathore3
1Resident, Department of Pathology, HCG Hospital, Bengaluru, Karnataka, India,
2Professor, Department of Pathology, Dr. S.N. Medical College, Jodhpur, Rajasthan, India,
3Resident, Department of Pathology, Dr. S.N. Medical College, Jodhpur, Rajasthan, India
Address for correspondence:
Dr. Kunal Sharma, 39, Bhairon Villas, Near Sardar Club, Ratanada, Jodhpur - 342 001, Rajasthan, India. Phone: +91-9784588630. E-mail: [email protected]
A case of 6-year-old male child with multiple, diffuse plaque-like lesions with crusting on the extremities, face, and the trunk. Histopathology of the lesions confirmed cutaneous chromoblastomycosis which is a slowly progressive mycosis caused by dematiaceous fungi. Sclerotic bodies highlighted by special stain were seen. The case was successfully managed for the same.
KEY WORDS:Pancreatitis, alcoholic, amylases, lipase, bilirubin
Chromoblastomycosis is a chronic, non-contagious, localized fungal infection of cutaneous and subcutaneous tissues. It is characterized by verrucous, crusted, or ulcerated lesions occurring mainly on the lower extremities and rarely on the face and other sites of the body. This mycosis is distributed worldwide, but most cases have been reported from tropical and subtropical regions.[1] It is especially common among bare-footed farm workers following traumatic implantation of dematiaceous fungi present mainly in soil, decaying vegetation, and rotting wood.[2,3] Diagnosis is made by direct microscopic demonstration of pathognomonic, brown sclerotic bodies (also called as Medlar bodies, muriform cells, copper pennies) in biopsy specimen and positive fungal culture for demonstration.[4,5] The present case deals with an atypical presentation in a child.
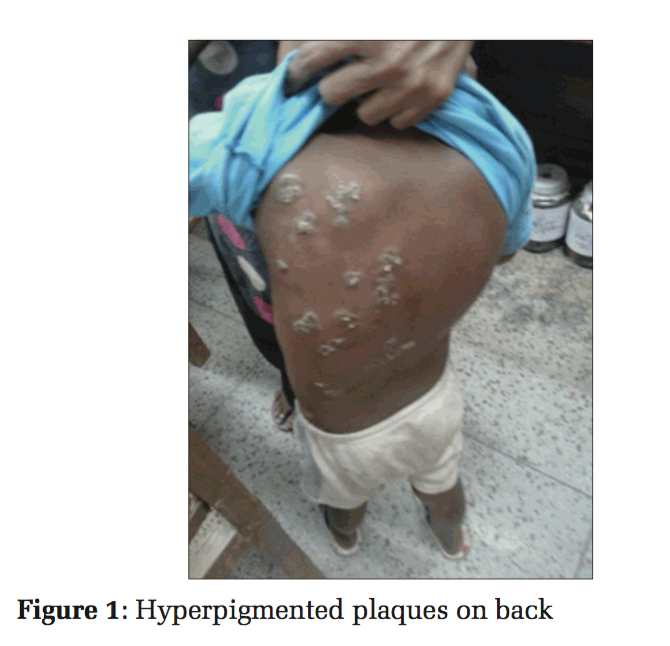
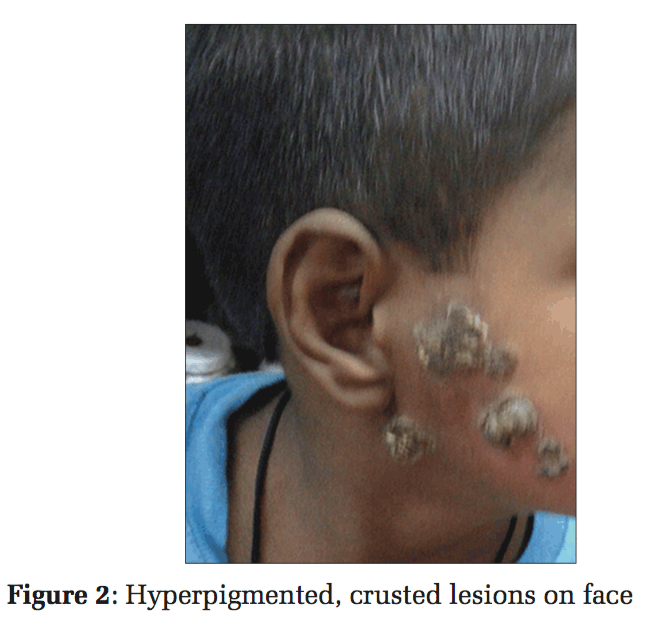
A 6-year-old boy, from Jodhpur district of Rajasthan, India, presented with slowly spreading hyperpigmented, crusted, verrucous plaques with the largest measuring 5 cm × 4 cm. The lesions started from the left arm and spread diffusely to involve all the extremities and later even the face and trunk (Figures 1 and 2).
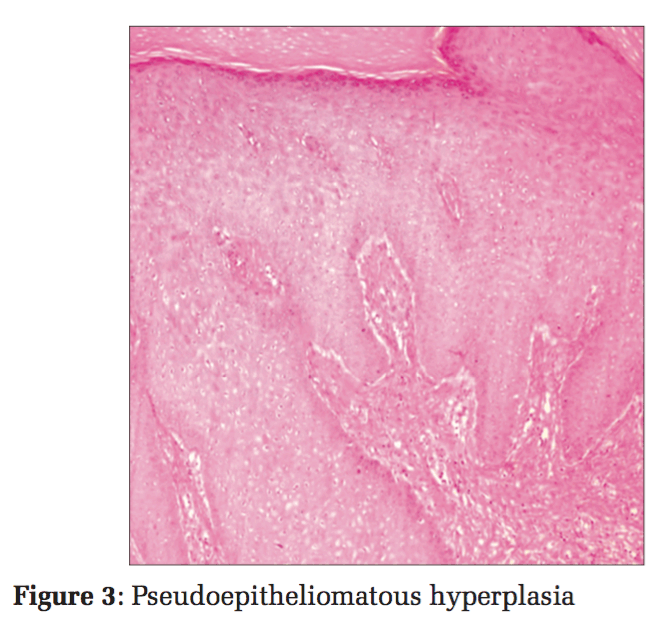
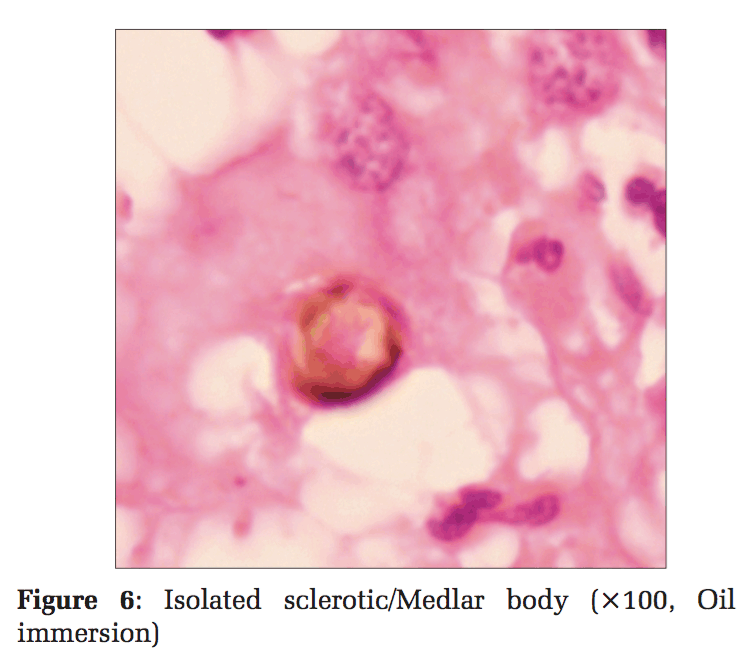
He did not have similar lesions in the past, and there is no significant history of any chronic illness or perinatal illness in the past. Skin biopsy samples were taken and subjected to histopathological examination. On histopathological examination, the section showed pseudoepitheliomatous epidermal hyperplasia with the presence of pigmented spores arranged singly and in chains with peripheral neutrophil infiltration (Figures 3 and 4). Reticular dermis showed mixed inflammatory infiltrate comprising of lymphocytes, macrophages, and plasma cells. Also seen are ill- defined granulomas along with golden-brown, thick-walled, spherical bodies about 6-12 μ in size suggestive of copper-penny/sclerotic bodies/Medlar bodies of chromoblastomycosis (Figures 5 and 6).
Ziehl–Neelson staining did not reveal any acid-fast bacilli. Periodic Acid-Schiff (PAS) stain highlighted the fungus. Fungal culture was not done. Sclerotic bodies and histopathological features such as pseudoepitheliomatous hyperplasia, along with PAS positivity, highlighting the fungus, were considered sensitive and specific enough to give a diagnosis of cutaneous chromoblastomycosis. The patient was treated for the same and the lesions have subsided.
This seems to be one of the rarer presentations of extensive involvement of the skin at various sites probably through hematogeneous transmission.
Cutaneous chromoblastomycosis, also called as “cladosporiosis,” “Pedroso’s disease,” and “Fonseca’s disease,” is a chronic cutaneous mycotic infection. The causative agent is dematiaceous fungi such as Fonsecaea pedrosoi, Fonsecaea compactum,
Phialophora verrucosa, and Cladophialophora carrionii. On rare occasions, Exophiala spinifera and Wangiella dermatitidis have also been implicated as the causative agents.[6] These fungi are saprophytes commonly found growing in soil, decaying vegetation, or rotten wood in subtropical and tropical countries. Madagascar, an island country off the coast of southeast Africa, has the highest incidence of chromoblastomycosis in the world.[7,8] The most common mode of infection is thought to arise as a result of traumatic implantation of the fungus into the skin. Hematogenous dissemination is another rare mode which may cause extensive cutaneous lesions. Men in the age group of 30-40 years are most commonly affected since they are most commonly involved in agricultural work and are prone to injuries.
Also known as “chromomycosis,” “cladosporiosis,” “Fonseca’s disease,” “Pedroso’s disease,” and “verrucous dermatitis,” it was first reported in Brazil, in 1914, by Max Rudolph, a German physician.[6,7] In 1915, Lane and Medlar described the sclerotic cells, which were subsequently called Medlar bodies.[4,5] The International Society for Human and Animal Mycology, in 1992, recommended the name chromoblastomycosis to define the disease, which Terra et al. coined in 1922. In India, it was first reported, in 1957, by Thomas et al., who reported two cases from Assam.
The disease is transmitted through inoculation of soil or vegetable matter contaminated by fungi or traumatic injury with wood splinters or thorns. The skin lesions generally arise on the lower extremities as a warty nodule and progress to form verrucous plaque-like lesions which may become tumorous and even cauliflower-like in appearance.[7] While some of the lesions heal with scarring, new lesions may appear in the vicinity resulting from the spread of the fungus along superficial lymphatics. Hematogenous spread is rare and carries a relatively grave prognosis.
Chromoblastomycosis must be differentiated from other mimickers such as leprosy, mycetoma, cutaneous tuberculosis, dermal leishmaniasis tertiary syphilis, phaeohyphomycosis, lobomycosis, paracoccidioidomycosis, and sporotrichosis. On histomorphology, a triad of pseudoepitheliomatous hyperplasia, marked neutrophilic infiltration with microabscess formation, and hallmark Sclerotic bodies are highly specific of chromoblastomycosis. The absence of characteristic globi, nerve involvement, caseation, lymphocytic infiltration, and histiocytes containing organisms or organism “grains” helps to rule out the differentials mentioned. There are no serological tests available and a combined histopathological and mycological diagnosis is a highly sensitive approach. Sclerotic bodies have been observed in 80-85% cases, and culture has been found to be positive in 70-75% cases as reported from the world over.[8] The common complications include lymphatic disruption with elephantiasis. Epidermoid carcinoma may occur rarely in long-standing cases.
There are several treatment options available including medication, cryotherapy, and surgery. Flucytosine with or without thiabendazole, itraconazole, terbinafine, and potassium iodide have all been used successfully for 6 months to 1-year duration. Pulse therapy is more economical with better patient compliance although optimal treatment duration depends on individual cases. Cryotherapy with liquid nitrogen has shown good results with minimal side effects and is recommended for small, localized lesions. In refractory cases, 5-aminolevulinic acid and irradiation in combination with antifungal therapy have been successfully used.[7]
Our case is unique because it is in a child of 6 years which is not the ideal age group. Furthermore, there are diffuse cutaneous lesions which suggest a rare form of hematogenous dissemination.


A diagnosis of chromoblastomycosis should always be considered as a differential in patients presenting with chronic skin lesions. Our case report reiterates the need to keep the atypical presentation of chromoblastomycosis such as diffuse skin lesions in mind. It also highlights the importance of coordination among clinicians and pathologists for the diagnosis of this slowly progressive, relatively refractory, cutaneous mycosis.Earlydiagnosisoftheselesionscanalso prevent the long-standing, rare complication of epidermoidcarcinoma.
We wish to thank all the patients who were part of the study, the Institution, the Department of General Surgery, the Department of Radiology and the Laboratory Unit of Adichunchanagiri Hospital and Research Centre, for their assistance, advice, help, and support.
Subscribe now for latest articles and news.