Introduction
SCD (Sickle cell disease) is characterized by abnormal morphology of red blood cells (RBC) as seen on the stained blood smear under the microscope 1. In the year 1910 Herrick first described the sickle shaped RBC 2. SCD is the most commonly encountered single gene disorder presenting as hemolytic anemia. 3 SCD is caused by a point mutation in the β-globin gene that resultsin a single nucleotide substitution, changing glutamic acid (GAG) to valine (GTG) in the sixth codon, and subsequently produces abnormal sickle hemoglobin. This disease is common in the tribal population of central India includes Andhra and Telangana. The frequency of sickle cell gene in Indian population varies from 2-3.5 % 4. It is hemolytic anemia that presents with an intervening crisis like a hemolytic crisis, vasoocclusive, sequesterative, aplastic crisis etc.
Though Hemoglobin electrophoresis and gene diagnosis are the gold standards for diagnosis of SCD, Peripheral smear examination remains an invincible tool in preliminary diagnosis in unsuspected cases, those in sickle crisis or those in hydroxyl urea. Sickling can be classified as Reversible sickling (RS)
Materials and Methods
The study was a prospective crossectional descriptive study conducted at a tertiary paediatric referral hospital. Thirty previously diagnosed cases of sickle cell disease, on regular follow up, admitted into the paediatrics department were included in the study. All the cases were previously diagnosed through high-performance liquid chromatography (HPLC) and genetic diagnosis. Very sick children and those with sickling crisis were not considered for the sickle cell index estimation. Cases of hemolytic anemia including sickle cell anemia without HPLC diagnosis were excluded from the study. Thirty age matched controls admitted in the hospital with non- hemolytic anemias were used as controls. The study period was 6 months.The blood drawn for routine screening of the patients at the time of admission was used in the study. No blood sample was drawn for the study separately in the patients or the controls. Informed consent was however taken from the parents for participation in the study. Venous blood collected in EDTA vacutainer was used. In order to avoid external factors influencing sickling, the patient was comfortably seated and a tourniquet was used. Blood collection was done with extreme care to see that there were no air bubbles. The sample processing and slide preparation was completed in 30 minutes. The smears were prepared by a senior technician and stained using ready made Leishman stain (Nise company) by manual method of staining.
Both reversible and irreversible sickle cells were quantified in 100 microscope fields and the total number of cells per field was derived.
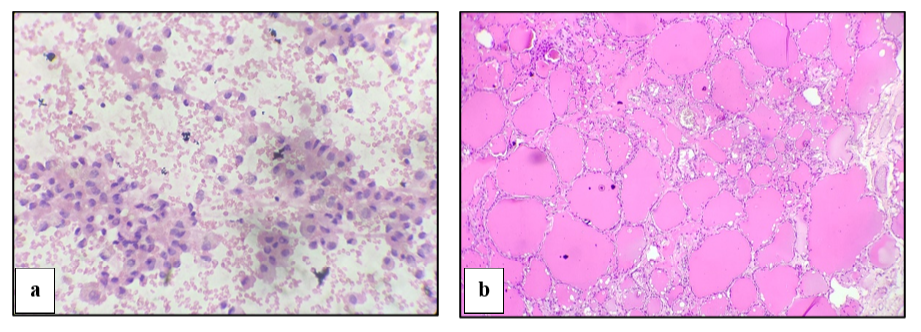
The smears were evaluated by two senior pathologists and the results given were in concordance. The photomicrographs of 10 oil immersion fields of the smear were taken and the images were transferred onto a computer and counting of red blood cells (RBC) was done under magnification using zoom in (+) option in Microsoft images (Figure 1).

Sickle cell index (SCI) was thus calculated in all 30 cases and controls.SCI is the mean percentage of the number of reversible and irreversible sickle cells in ten 100 X microscope fields.
Sickling Index = X/ Y
x = Total number of sickle RBC in 10 oil immersion fields
y = Total number of RBC in 10 oil immersion fields
Also, patient related information was collected from the hematology records which included age, gender, sickle cell genotype, associated complications at the time of admission. Treatment history with transfusions or hydroxyurea was also recorded.
We counted both reversible and irreversible sickle cells
Reversible sickle cells were defined as normochromic cells with holly leaf splinter, boat envelope, or ellipsoid configuration.
Irreversibly sickled cells were defined as hyperchromic cells, length twice than width crescent with elongated and pointed ends.
Results
Out of the 30 patients in this study 7 (23.33%) were in the less than 2 years age group and most of them 23 (76.66%) were more than 2 years old. The male to female ratio was almost equal with a slight female preponderance. The distribution of cases according to age, sex, and genotype is given in Figure 2. HbSS variant or homogenous SCD was the commonest cause of admission and constituted 21(70%) of the cases. Non-SS variants were seen in 9(30%) of the cases.

Comparison SCI among cases and controls is given in Table 1. The comparison of SCI in SS and Non-SS genotype has also been tabulated in Table-1. The sickle cell index in cases is noted to be higher than in normal children.
Mean SCI was notably higher in patients with age more than 2 years, SS genotype, and those needing treatment with a transfusion or hydroxyurea as given in Table 2
|
Patients |
Mean Scikle cell index |
Range |
|
Controls N=30 |
0.12 |
0.0 - 0.22 |
|
Cases N=30 |
3.04 |
0.19 - 5.8 |
|
SS genotype N=21 |
3.75 |
0.22 - 5.8 |
|
Non-SS genotype N=9 |
1.84 |
0.19- 2.4 |
|
Category |
Mean Sickle cell index |
|
Age <2 years SS + non-SS (N = 7) |
1.79 |
|
Age ≥ 2years SS only (N = 17) |
3.78 |
|
Age ≥ 2 years non-SS only ( N = 6 ) |
1.78 |
|
Hydroxyurea treatment (N = 8) |
3.92 |
|
On Transfusion (N = 16) |
3.83 |
Discussion
Sickle cell disease is the most common monogenic disease associated with recurrent episodes of acute illness and multisystem disease.SCD was first described by Herrick in 1910 in a West Indian patient with anemia and heart murmurs from Chicago 5. After he published his paper describing the sickle shaped erythrocytes. Pauling identified the abnormalities of hemoglobin in SCD and coined the term “molecular disease” in 1949. 4
In 2010 the number of newborn children with SCA was 30, 5800 globally and a statistical model by Piel et al suggested that it will be around 4 times by mid 21 century. 5 The subtype of SCD in India is similar to the Arab Indian haplotype. 6
The sickle cell gene in India was first described in the Nilgiri hills of northern Tamilnadu in the year 1952. 7 Sickle gene is widely reported among the people of the Deccan Plateau of Central India 8 which is where Hyderabad is located. The present study was conducted at a tertiary children's referral hospital in this city. Studies of the anthropological survey of India show the incidence and distribution of sickle cell gene are as high as 35% in some communities in this part of the country. 8
The sickle cell mutation affects the Beta chain of adult hemoglobin which causes the sickle Hemoglobin (Hb). Single Hb S gene results in the sickle cell trait which has a relatively benign course. Inheritance of the HbS gene from both the parents results in Homozygous sickle cell disease which is a severe condition causing rapid hemolysis, painful and serious complications. Hb SS is the most common form of hemoglobinopathy. It has an inheritance of sickle gene from both the parents, other disorders of SCD result from coinheritance of the sickle gene.
HbS is different from usual Hemoglobin A because there is only a single nucleotide change. (GAT to GTT) and this results in the substitution of valine for glutamate at the 6th position of the beta chain of the hemoglobin molecule. This results in abnormalities of polymerization upon deoxygenation and this leads to sickling of the red blood cells. Polymerization results in a permanently altered membrane protein. Sickled RBC s are usually removed by the spleen. Hemolysis occurs after sickling. In HbSS membrane abnormality is significant with an increased influx of calcium into the cell. The ionized calcium in HbSS is twice normal to up to 4 times normal. HbS undergoes polymerization under hypoxic conditions or acidic Ph. Sickling is caused by low Ph increased 2, 3 DPG or low oxygen tension and dehydration.
The molecular basis of sickling has been deciphered with lowered oxygen tension when a continuum of changes begins to occur. It progresses to the formation of an orderly polymer. From a lesser amount of polymer to a higher amount after severe deoxygenating, accumulation of this intracellular polymer occurs. Its amount and alignment determine the degree of loss of flexibility. Sickling causes RBC to occlude the vessels and this vasoocclusion further causes further hypoxia. Upon reoxygenation, sickle cells return to normal. Repeated cycles of sickling and unsickling lead to hemolysis and subsequent clinical symptoms.
The concept of Reversible (RS) and Irreversible sickling (IRS) has also been studied by many researchers.IRS results from damaged RBC membrane due to repeated sickle hemoglobin polymerization 9
Diggs & Bibb have reported ISC as a hallmark of sickle cell anemia 10. Correlation of IRS to disease symptoms has also been attempted. Morphological description of IRS includes sickle cells with an axial ratio greater than 2:1 and a pointed angle 11. These criteria were established by different studies after understanding the irreversibility of sickle RBC on reoxygenation cells that retain their sickle shape after oxygenation is called IRS and those that return to their normal shape are called RS cells. Studies did not find any correlation between vasoocclusive crises and increased IRS 12 others found the increased numbers of RS cells to correlate with sickle cell crises 12, 13. Zipursky, in his study, did not find any correlation between the number of sickled cells or the degree of sickling to sickle cell crisis.
Sickling of RBC as discussed in detail is the hallmark of SCD. However, the variation in the degree of sickling has been a matter of interest to the pathologists. Although the final diagnosis of a complicated SCD case does not rest in peripheral smear examination. Quest for a role of their quantification in determining the disease severity or type of genomic abnormality has always been an interesting prospect.
Many other parameters like reticulocyte count, hemoglobin, white cell count have been studied in patients with SCD with inconsistent results. The presence of increased numbers of deformed and sickle cells in the peripheral smear and its relation to the patient's demographic and clinical profile was the main objective of our study.
We used the method described by Alvarez et al, 14 and found that it was indeed possible to perform a quantitative analysis of sickle cells. On comparing the mean sickle cell index our study found a significantly higher index in cases as compared to normal controls (0.12%). This small percent could be artefactual changes in the RBC in normal smears. Alvarez et al, in their similar study, found 0.28% of control smears showing sickle cells. Their SCI of subjects was 4.59% in comparison to 3.04% in our study. Their mean SCI for SS genotype was 5.37 while it was 3.75 in our study. We noticed a higher mean SCI in patients who are older than 2 years. The sickling index was higher in patient son treatment with hydroxyurea treatment and those with a history of transfusions. These results do not correspond to the findings of Alvarez et al who found a lower sickling index in patients who were on treatment. Their study was in adult patients while we had pediatric subjects in our study, and we collected the samples at the time of admission.
This relationship of age and sickling is because of the persistence of fetal hemoglobin (HBF) in infants and very young children. Studies have revealed that HbF usually disappears from the red blood of infants after about 6 months. However, the exact time of disappearance of HbF may vary and the signal that determines the switch from fetal to adult hemoglobin is not known. Edoh in their study found that HbF was higher nearly 98%, at birth and decreased at a rate of about 10% per fortnight till about 6 months when it disappeared completely. 15 This waning off of HbF is due to the switching from fetal to adult hemoglobin in infancy. 16 They also found that HbF is higher in adults with sickle cell disease than in non-sickle cell subjects. Therefore, the younger children had a lesser sickling index.
The sickling index was higher as expected in SS genotype which is the homozygous variant and lower in the non-SS variants as it is known that patients with non-SS variants have HBA also which causes a lower rate of sickling. Okwi in their study in which they compared different methods for diagnosis of sickle cell genotype found that the peripheral film method had high specificity, low sensitivity in being able to predict SS from other non-SS variants as it showed a higher rate of sickling. The other test modalities they used were sickling and solubility tests and peripheral blood film method. 17. A higher sickling index may also help us to suspect and predict SS genotype in a new undiagnosed case of SCD before HPLC.
The treatment of SCD includes two types of strategies one targeting the relief of symptoms and the other prevention of symptoms. For immediate relief of symptoms blood transfusions, analgesics, antibiotics are given. For symptom prevention one of the main treatment approaches is the induction of fetal hemoglobin mainly with the drug hydroxyurea. Since individuals with SCA produce HbS instead of HbA, HbS polymerization leads to symptoms of SCA 18 . Fetal hemoglobin inhibits HbS polymerization by directly interfering with polymerization and by reducing the concentration of HbS production 19 . Hence, the elevation of HbF levels ameliorates the severity of SCA 19
Most of the children receiving transfusions or hydroxy urea treatment as discussed previously had a higher sickling index suggesting more severe disease.
The study finds the sickling index an interesting simple tool to suggest increasing or decreasing hemolysis. It may also be useful in predicting the prognosis in patients on treatment and follow up. More studies with larger samples will provide more insights
Conclusion
Quantification of sickle cells on the peripheral smear is very simple and easy to perform. A higher index helps in predicting a more severe disease and homozygous phenotype. This study found a higher index in older children. Possible reasons for the increased sickling in older children could be erythroid in origin, lower HbF, increasingly defective membrane, vascular in origin, progressive vasculopathy, or decreased clearance from circulation due to impaired spleen function. On expected lines, the study finds a higher sickling index in SS genotype in comparison to non-SS genotype. Patients needing treatment also showed a higher sickling index. These findings can be used as a simple tool in objectively assessing the SCD genotype in new cases before HPLC or genotyping. Patients requiring treatment with hydroxyurea or transfusions can also be suggested by high sickling index which is also a finding in our study.
Conflicts of interest
The authors report no conflicts of interest