Journal of Medical Sciences and Health
DOI: 10.46347/jmsh.v9i3.23.189
Year: 2023, Volume: 9, Issue: 3, Pages: 324-330
Case Series
Kenza Dafir1,2, Fatima Zahrae Bouzid1,2, Maria Mansouri1 , Hassan Akallakh1,2 , Imane Ait Sab3,2, Mohammed Bouskraoui4,2, Nisrine Aboussair1,2
1Genetics Department, Clinical Research Center, University Hospital Center Mohammed VI, Marrakech, Morocco,
2Faculty of Medicine and Pharmacy, Cadi Ayyad University, Marrakech, Morocco,
3Pediatric department B, mother and child hospital, University Hospital Center Mohammed VI, Marrakech, Morocco,
4Pediatric department A, mother and child hospital, University Hospital Center Mohammed VI, Marrakech, Morocco
Address for correspondence:
Kenza Dafir, Genetics Department, Clinical Research Center, University Hospital Center Mohammed VI, Marrakech, Morocco, Faculty of Medicine and Pharmacy, Cadi Ayyad University, Marrakech, Morocco.
E-mail: [email protected]
Received Date:25 May 2023, Accepted Date:11 September 2023, Published Date:28 December 2023
Pycnodysostosis is a rare bone dysplasia with an incidence of 1 in 100,000 cases. It is a genetic disease (from the Greek pycnos = dense, dys = trouble and osteon = bone) also called osteochondrodysplasia, first described in 1962 by Maroteaux and Lamy. It is due to a dysfunction of the osteoclasts, caused by a mutation in the gene that codes for the cathepsin K enzyme. We highlight the role of the geneticist in the early diagnosis and management of pycnodysostosis as well as in the elaboration of an adequate genetic counseling based on a series of 14 clinical cases.
Keywords: Pycnodysostosis, Atypical Fracture, Osteoclast, Cathepsin K, Facial Dysmorphia
Pycnodysostosis (OMIM: 265800) also called Toulouse Lautrec disease is a lysosomal storage diseases, inherited in an autosomal recessive manner, due to osteoclastic dysfunction 1. The first case was described in 1923 by Montanari, but the disease as we know it today was described by Maroteaux and Lamy in 1962 2, 3. The prevalence of pycnodysostosis is one case per 1.7 million inhabitants, with a sex ratio of 1 2. This disease is related to a deficiency of cathepsin K, which is a protease of the cysteine protease family present in osteoclasts. Several mutations in the CTSK gene reduce its expression leading to increased bone density 4. We report in this work the first series of 14 cases of pycnodysostosis collected within the genetics department, clinical research center, University Hospital Mohammed VI of Marrakech and we take stock of this rare genetic syndrome.
We report the first series of 14 clinically and radiologically confirmed cases of pycnodysostosis. All patients were referred to the medical genetics department of Mohammed VI University Hospital in Marrakech for bone fragility and statural delay. The series was conducted over a seven-year period from January 2013 to January 2020.
The inclusion criteria for our study comprise the presence of clinical signs indicative of pycnodysostosis, including characteristic facial dysmorphism, typical bone involvement, and other associated symptoms, along with a diagnostic orientation supported by radiological examinations revealing the characteristic lesions of pycnodysostosis. However, some patients were excluded from the study based on specific criteria, including the presence of another major genetic disease that could potentially impact the observed results or symptoms, as well as insufficient medical data and relevant information to
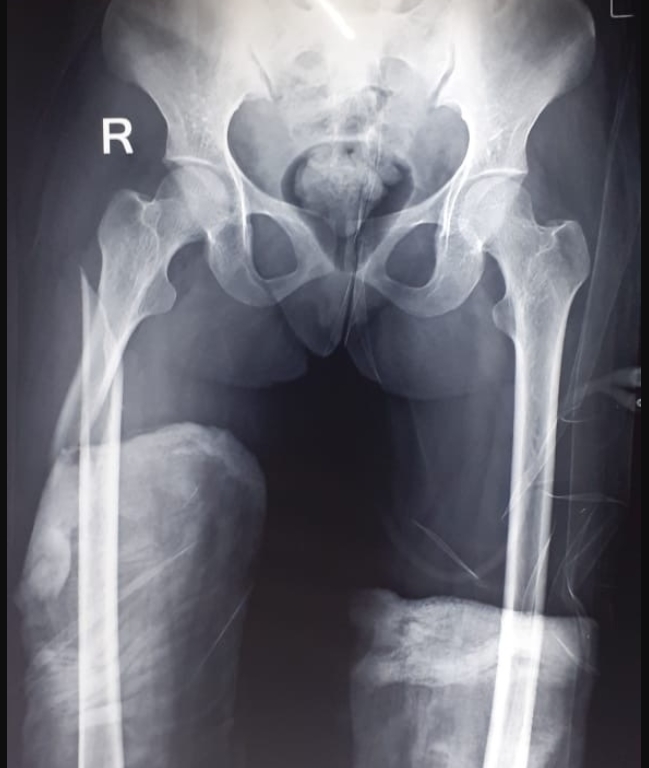
The sex ratio of our series was 10 males to 04 females, the age of our patients ranged from 03 to 51 years with a mean age at diagnosis of 12.14 years, consanguinity was reported in 05 patients. The clinical information of the 14 patients is summarized in Table 1, Table 2. Figure 1, Figure 2 shows phenotypes and X-rays examples of our patients.
|
|
Patient 1 |
Patient 2 |
Patient 3 |
Patient 4 |
Patient 5 |
Patient 6 |
Patient 7 |
|
Sex |
Female |
Male |
Male |
Male |
Female |
Male |
Male |
|
Age at diagnosis |
Five years |
Four and a half years old |
Four and a half years old |
51 years |
Five years |
24 years |
11 years |
|
Maternal age |
40 years |
42 years |
30 years |
deceased |
26 years |
44 years |
42years |
|
Craniofacial dysmorphia |
- Skull deformation, open anterior fontanel - Frontal hump - Exophthalmos - Prominent nose with wide base - Big ears -Full cheeks - Short and marked Phyltrum - Drooping wedge mouth - Retrognatism |
- Skull deformation, open anterior fontanel - Frontal hump - Exophthalmos - Hooked nose - Drooping cheeks - Long Phyltrum - Thin upper lip - Small mandible |
- Skull deformation -Frontal and occipital humps - Exophthalmos - Small mandible with retrognatism - Prominent nose with wide base - Dental anomalies |
-Skull deformation -Frontal and occipital humps -exophthalmos -small mandible with retrognatism -Prominent nose with wide base - dental anomalies |
- Skull deformation, open anterior fontanel - Frontal and occipital humps - Oblique palpebral slits at the bottom, exophthalmos - prominent nose - Curved cheeks, reduced average floor height - Small mandible dental anomalies |
-Skull deformation -frontal hump - Exophthalmos - Prominent nose with wide base - Small mandible with retrognatism - Poor oral health, dental abnormalities |
-bulky skull - frontal hump - Exophthalmos - Prominent nose - Drooping cheeks - Micrognathir - Dental abnormality, sharp teeth |
|
Associated deformities |
-Stocky short hands -Brachydactyly -short phalanges -inch very wide -Small feet, -short toes -Curved fingernails |
-Small, square hands -Brachydactyly -short phalanges -Small feet, - short toes -Dysplastic fingernails |
- Small and square hands, - Brachydactyly - Short phalanges - Small feet, - Short toes -Dysplastic nails |
-Small, square and aged hands -Brachydactyly - Short phalanges - inch very wide - Small feet, short toes - Curved fingernails |
- Small and square hands, - Brachydactyly -short phalanges -Small feet, short toes -Dysplastic fingernails |
-Small, square hands -Brachydactyly -short phalanges -Small feet, -short toes -Dysplastic fingernails |
-Small, square hands -Brachydactyly -short phalanges -wide thumb -Small feet, -short toes -Irregular fingernails |
|
Antecedent Fractures |
00 fracture |
00 fracture |
00 fracture |
04 fractures |
00 fracture |
06 fractures |
01 fracture |
|
|
Patient 8 |
Patient 9 |
Patient 10 |
Patient 11 |
Patient 12 |
Patient 13 |
Patient 14 |
|
Sex |
Male |
Female |
Male |
Male |
Male |
Female |
Male |
|
Age at diagnosis |
Three years |
Six years |
Five years |
four years |
21 years |
15 years |
seven years |
|
Maternel age |
33 years |
42 years |
29 years |
33 years |
49 years |
47 years |
34 years |
|
Craniofacial dysmorphia |
-Skull deformation, open anterior fontanel -frontal hump -Exophthalmos -Prominent nose -Poor dental implantation -Retrognatism |
-bulky skull, open anterior fontanel -frontal hump -Exophthalmos -Prominent nose -Drooping cheeks -Long Phyltrum -Small mandible |
-Skull deformation, open anterior fontanel -Frontal and occipital humps -Exophthalmos -small mandible with retrognatism -Prominent nose with wide base -dental anomalies, sharp teeth |
-Skull deformation, open anterior fontanel -frontal hump -Exophthalmos -Prominent nose -dental abnormalities |
-Skull deformation -Frontal and occipital humps -Bitemporal shrinkage -Down oblique palpebral slits -exophthalmos -Hooked nose -Drooping cheeks -micrognathia -dental anomalies |
-Skull deformation -frontal hump -Exophthalmos -Prominent nose with wide base -Big ears -Small mandible with retrognatism -Poor oral health, dental abnormalities |
-Skull deformation, open anterior fontanel -Frontal and occipital humps -exophthalmos -Micrognathia -Prominent nose with wide base -sharp teeth with poor dental implantation |
|
Associated deformities |
-Small and square hands, -Brachydactyly -short phalanges -Small feet, -short toes -Dysplastic nails |
-Small and square hands -Brachydactyly -Small feet, - short toes -Small nails |
-Small and square hands, -Brachydactyly -short phalanges -very large thumb -Small feet, -short toes -Irregular nails |
-Small and square hands, -Brachydactyly -short phalanges -Small feet, -short toes -Dysplastic nails |
-Stocky short hands -Brachydactyly -short phalanges -Small feet, -short toes -Dysplastic, cracked nails |
-Small, square hands -Brachydactyly -short phalanges -very large thumb -Small feet, -short toes -Dysplastic nails |
-Small, square hands -Brachydactyly -short phalanges -Small feet, -short toes -Dysplastic nails |
|
Antecedent Fractures |
01 fracture |
00 fracture |
00 fracture |
01 fracture |
03 fractures |
02 fractures |
00 fracture |


The genealogical study and the clinical signs, including a characteristic facial dysmorphia, as well as the radiological examination led to the diagnosis of pycnodysostosis. DNA extraction was performed, for a possible molecular study to confirm the genetic diagnosis.
Pycnodysostosis is an inherited condensing osteopathy transmitted in an autosomal recessive manner 1. Consanguinity is described in 30% of cases 5, which is close to the consanguinity rate (35.7%) found in our series.
This disorder is caused by a homozygous mutation or a composite heterozygous mutation in the gene that codes for the cathepsin K enzyme (CTSK), located on chromosome 1q21. The cathepsin K enzyme is a lysosomal cysteine protease highly expressed in osteoclasts, the cells responsible for bone resorption, and is involved in the degradation of bone matrix proteins. It plays a crucial role in enzymatic activity essential for normal bone remodeling In individuals with pycnodysostosis, mutations in the CTSK gene disrupt the structure or function of the enzyme, resulting in enzyme dysfunction. This alteration leads to an abnormal accumulation of bone matrix proteins, particularly type I and II collagens, osteopontin, and osteonectin. Because of the accumulation of these proteins, osteoclasts exhibit impaired bone resorption function, leading to increased bone density and characteristic abnormalities, such as bone fragility, frequent fractures, and short stature. To date, 45 different mutations in the CTSK gene have been reported, encompassing nonsense, missense, frameshift, and splice site mutations, as well as small deletions, and small and large insertions (Alu sequence). This genetic diversity contributes to the phenotypic variability observed in individuals with pycnodysostosis, with clinical manifestations varying in severity from one patient to another. The detection of mutations not only confirms the diagnosis but also enables prenatal molecular diagnosis 6. Additionally, it opens avenues for understanding pathological mechanisms and provides opportunities for the development of new targeted therapeutic strategies in the future.
The clinical implications of the identified genetic mutations may have a significant impact on the management and prognosis of patients with pycnodysostosis, as well as a more precise genetic counseling. Molecular diagnosis is not done in Morocco. we carried out DNA extraction for further research studies. The diagnosis is currently based on genealogical, clinical and radiological criteria.
Pycnodysostosis is characterized by cranial and clavicular dysplasia, total or partial dysplasia of the distal phalanges, and generally high bone density7. Additionally, individuals with pycnodysostosis may present with maxillary and mandibular hypoplasia, an open, and sometimes absent mandibular angle, delayed eruption of permanent teeth, impacted teeth, and severe dental crowding with a higher prevalence of caries and periodontal abnormalities 8, 9. The results of our study align with those reported by Maroteaux P. et al.7, Bléno K et al. 8 and Fonteles CSR et al. 9, as all our patients exhibited one or more of the described bone anomalies, and 11 out of 14 patients had dental anomalies and caries.
Pycnodysostosis is characterized by cranial and clavicular dysplasia, total or partial dysplasia of the distal phalanges and generally increased bone density 7, as well as maxillary and mandibular hypoplasia with an open and sometimes absent mandibular angle, delayed eruption of permanent teeth, impacted teeth, and significant tooth crowding 8. The prevalence of caries and periodontal anomalies is also higher 9.
Pycnodysostosis is typically diagnosed at an early age through the combination of short stature, late closure of the fontanelles, and characteristic facial dysmorphism 10. Our clinical data, detailed in Table 1, Table 2, reveal that all of our patients exhibit these signs, aligning with observations reported in the literature by Nardi J. et al. The diagnosis may also be made at an older age, following pathological fractures resulting from minimal or moderate trauma due to severe bone fragility, as highlighted by Landa S et al. 11 and Ferguson JW et al. 12. Our results align with their studies, indicating that 04 out of 14 patients in our cohort were referred for bone fragility and experienced frequent fractures
We can summarize the characteristic signs of the syndrome of pycnodysostosis found in the literature in 13:
Short stature or nanism
Characteristic facial dysmorphia, namely a large head with presence of wormian bones, the persistence of the anterior fontanel, frontal bumps, exophthalmos, a prominent nose, micrognathia
Dental abnormalities
Dysplastic and cracked nails.
Our clinical data detailed in Table 1, Table 2 are similar to the literature, which allowed us to evoke this syndrome and to orient paraclinical examinations.
Radiographs of the hands, dorsolumbar spine, and skull of our patients align with the findings of Mandala k.et al. 14, Ramaiah KKK et al. 15, and Wolpowitz A et al. 16 revealing diffuse and increased osteocondensation at the base of the skull, dorsolumbar spine, and hands. Additionally, our observations include open fontanelles, wide sutures, and the presence of Wormian bones, along with hypoplasia of facial bones, lower jaw with the disappearance of the mandibular angle, and resorption of phalangeal extremities.
Differential diagnosis occurs mainly with osteopetrosis, cleidocranial dysplasia and idiopathic acro-osteolysis 5. Osteopetrosis is distinguished from pycnodysostosis by compressive neuropathies, hypocalcemia associated with secondary tetanus convulsions, and potentially fatal pancytopenia. It is due to mutations in at least 10 genes that have been identified to date and which are implicated in 70% of cases 17.
In cleidocranial dysostosis, patients present, unlike pycnodysostosis, a broad and flat forehead, hypertelorism and drooping narrow shoulders. It is caused by RUNX2 gene (6p21) mutations 18. Patients with idiopathic acro-osteolysis have short fingers or toes, associated with nail and skin abnormalities. It differs from pycnodysostosis by the absence of a characteristic facial dysmorphia 19. No specific treatment for pycnodysostosis is currently available. The therapeutic approach is essentially based on the prevention of fractures and caries. This is why regular follow-up by an orthodontist of patients suffering from pycnodysostosis is so important. The curative treatment is that of complications (fractures and tooth extractions). 70% of our patients have undergone at least one surgical procedure such as osteosynthesis. Orthopedic immobilization treatments are performed at least once in 40% of cases. In patients with growth hormone (GH) defective secretion, GH supplementation increases IGF-I concentration and improves linear growth in 33% of cases 20, 21. However, current data are insufficient to evaluate the efficacy criteria of GH supplementation. None of our patients have been able to benefit from this therapeutic component because the availability of GH in our context remains difficult.
Pycnodysostosis is a rare, clinically distinct, autosomal recessive genetic disease. Diagnosis is based essentially on clinical and radiological arguments, the disease is often revealed by fractures. Management is symptomatic and multidisciplinary.
Through this work, we highlight the role of the geneticist in the diagnosis and management of pycnodysostosis, as well as in the development of appropriate genetic counselling.
This study is the first estimate of the prevalence of pycnodysostosis in Morocco. In fact, between 2013 and 2020, 14 cases were identified. This prevalence confirms the rarity of the disease in the Moroccan population.
This study highlights that this pathology is still unknown by the majority of patients and their families. The diagnosis is established between 12 and 14 years of age, confirming the significant diagnostic delay. Moreover, one patient was diagnosed at an advanced age which is 51 years old.
The objective of this study is to sensitize the population as well as health professionals regarding this rare, unknown pathology. Thus allowing early treatment and improving the quality of life of patients.
The limited number of patients in our series as well as economic constraints and absence of a national registry do not allow us to extrapolate all the results to the Moroccan population.
The results of our work can serve as a starting point for further research with larger and more diverse samples, which would allow for stronger generalization of the results.
Subscribe now for latest articles and news.