Journal of Medical Sciences and Health
DOI: 10.46347/jmsh.2019.v05i03.008
Year: 2019, Volume: 5, Issue: 3, Pages: 44-47
Case Report
Archana A Randale1, Sanjay N Parate2, Saroj A Meshram1, Shilpa P Tathe1, Milind A Bhatkule1
1Assistant Professor, Department of Pathology, Government Medical College and Superspeciality Hospital, Nagpur, Maharashtra, India,
2Professor and Head, Department of Pathology, Government Medical College and Superspeciality Hospital, Nagpur, Maharashtra, India
Address for correspondence:
Dr. Archana A Randale, Department of Pathology, Government Medical College and Superspeciality Hospital, Nagpur - 440 003, Maharashtra, India. Phone: 91-9860130404. E-mail: [email protected]
Amyloidosis is a wide spectrum of disease characterized by extracellular deposition of misfolded protein which has common morphological, structural, and staining properties but differs in their protein composition. Hepatic amyloidosis can present as clinicoradiological dido and requires liver biopsy and ancillary histopathological techniques not only to attest the diagnosis but also for typing of amyloidosis. Here, we report a case of hepatic amyloidosis with preserved liver function test despite having massive hepatomegaly.
KEY WORDS: AA type, amyloidosis, massive hepatomegaly.
Amyloidosis is a wide spectrum of disease characterized by extracellular deposition of misfolded protein that aggregates to form insoluble fibrils. These fibrils have common morphological, structural, and staining properties but differ in protein composition.[1] The worldwide estimated incidence of amyloidosis is 5–9 cases per million patients per year.[2] Practically no organ/tissues are spared from infiltration by amyloid fibrils. However, clinically dominant hepatic amyloidosis presenting as diffusely infiltrative disease in the form of massive hepatomegaly with preserved hepatic function is unusual.[3,4]
Here, we report a case of hepatic amyloidosis with preserved liver function test despite having massive hepatomegaly.
A 42-year-old female presented with history of dull aching, non-radiating pain in the right hypochondriac, and epigastric region with distension of abdomen for 2 months. Furthermore, she had history of loss of appetite and breathlessness for 1 month. There was no history of jaundice, itching, altered bowel habit, or altered sensorium. There was no history of major illness in the past. On examination, patient was conscious, oriented with vitals within normal limits. However, on per abdominal examination, liver was palpable 5 cm below the costal margin with smooth surface, rounded borders, and firm consistency.
Blood investigation was done showed normal levels of liver enzymes (alanine aminotransferase – 31 U/L, aspartate aminotransferase – 46 U/L, alkaline phosphatase–70U/L,andbiliru1.2 mg/dl)andkidney function test (blood urea nitrogen – 32 mg/dl and serum creatinine – 0.8 mg/dl). Hemogram revealed normocytic normochromic anemia with total white blood cells and platelet count within normal limits. Erythrocyte sedimentation rate was 20 mm of Hg. Triple H markers (HIV, HBsAg, and HCV) were negative.
With this clinical history and baseline investigations, the patient was subjected to computed tomography of abdomen which showed grossly enlarged liver with disproportionate enlargement of the left lobe of liver. Liver was measuring 21 cm craniocaudally. There was no evidence of any focal lesion in the liver [Figure 1]. Radiological diagnosis offered was diffusely infiltrative liver pathology. Upper gastrointestinal endoscopy was also done and did not show any significant pathology.
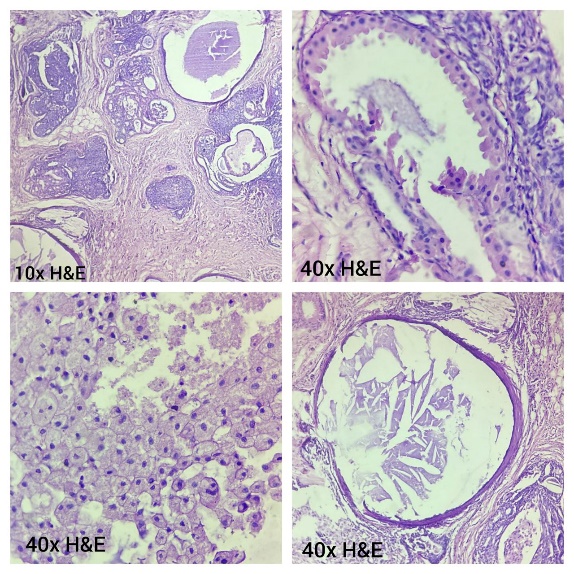
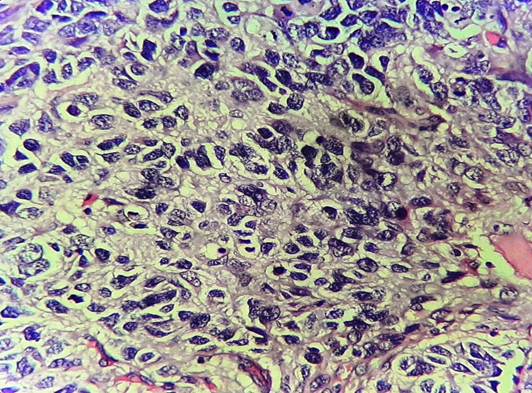
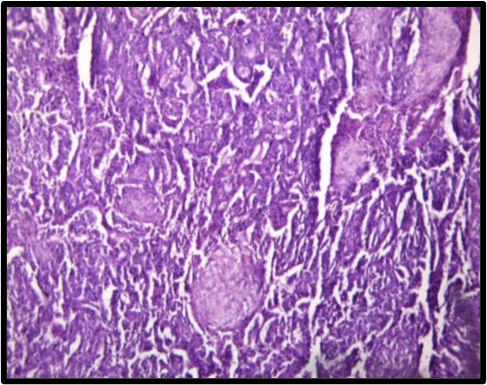
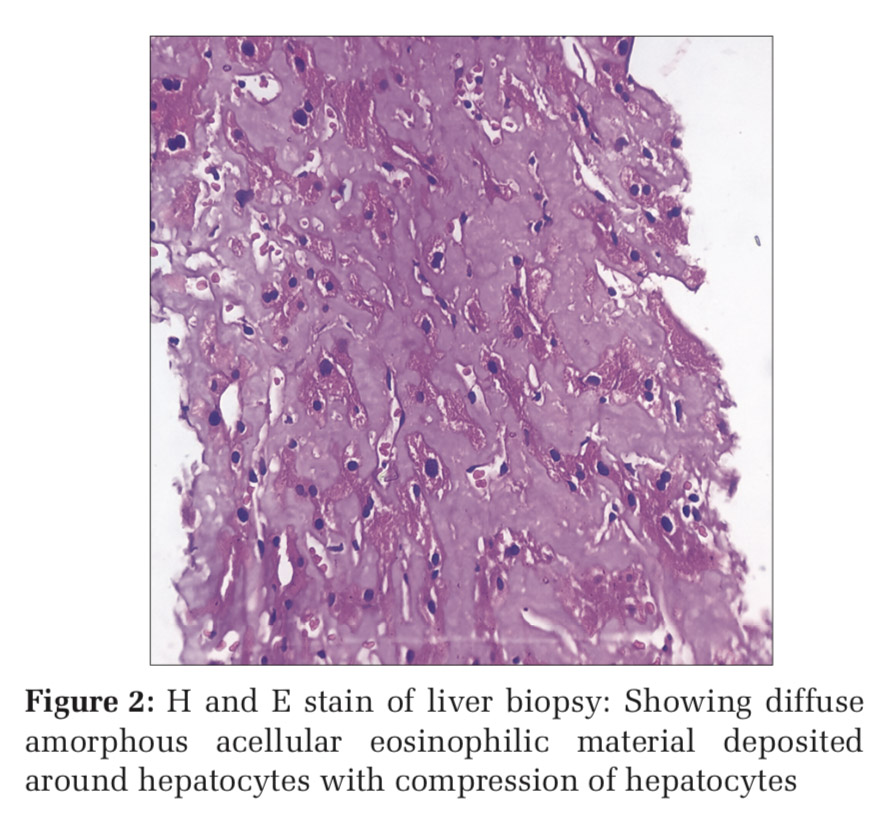
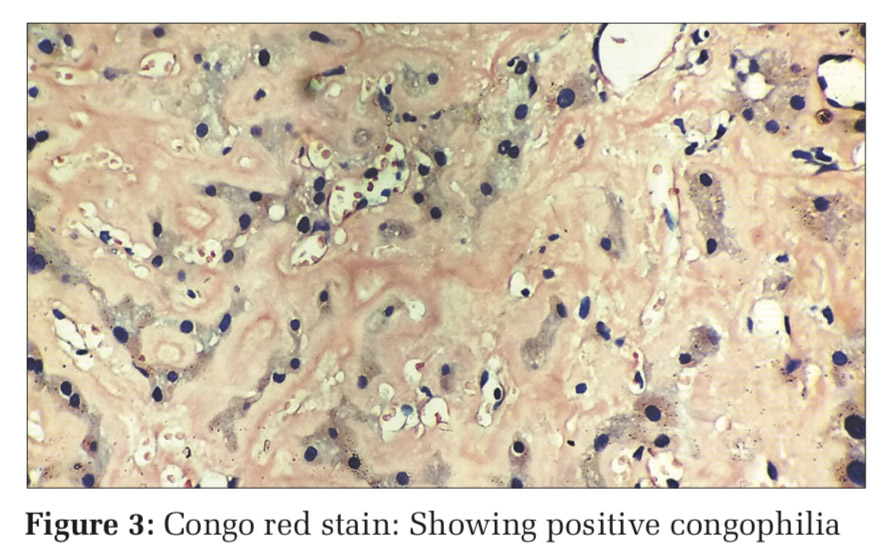
To overcome with this clinicoradiological camouflage, liver biopsy was done which showed deposition of amorphous acellular eosinophilic material between and around hepatocytes. Hepatocytes appeared compressed and atrophic at places [Figure 2]. With high suspicion for amyloid deposits on hematoxylin and eosin stain, Congo red stain was advised which showed positive congophilia [Figure 3]. Further to know the type of amyloid, immunohistochemistry was done [Figure 4]. It showed apple-green birefringence under polarized microscope and strong positivity for serum amyloid associated (SAA) protein. Thus, confirming the diagnosis of hepatic amyloidosis (AA type). Serum electrophoresis was also done and did not reveal any M band.
Due to her complaint of breathlessness, two- dimensional (2D) echo was done which showed features of the nonobstructive type of hypertrophic cardiomyopathy suggestive of some infiltrative pathology.
Amyloidosis is a wide spectrum of disease characterized by extracellular deposition of misfolded protein that aggregates to form insoluble fibrils. These fibrils have common morphological, structural, and staining properties but differ in protein composition. It is this protein deposited determines the type of amyloid.[1,5]
Two principle types of amyloidosis are:
Other rare types of amyloidosis include dialysis associated ß2 microglobulin-related amyloidosis and familial amyloidosis polyneuroapthy.[1]
At present, the AA/AL ratio is 1:17–1:38. This may be due to increasing recognition of myeloma associated amyloidosis and advanced treatment modalities for infectious/inflammatory diseases.[7]
Systemic primary AL amyloidosis mainly involves tongue, heart, gut, muscle, nerve, and skin whereas secondary amyloidosis involves liver, spleen, kidney, and adrenal gland predominantly.[8] Hepatic involvement is more common with secondary AA amyloidosis and presents as diffuse infiltration.[9]
Hepatic amyloidosis starts with deposition of amyloid in the space of Disse first, progressively encroaching on to adjacent hepatic parenchyma and sinusoids. As the disease advances, deformity, pressure atrophy, and disappearance of hepatocytes can occur resulting in total replacement of large areas of liver parenchyma. With this massive infiltration liver gets a rubbery, elastic consistency resulting in what is called as “lardaceous liver” appearance on cut surface.[10]
Clinically, these patients mostly have mild manifestations with preserved hepatic function despite severe liver encroachment.[11] Radiological findings of hepatic amyloidosis can be non-specific in the form of diffusely infiltrative hepatic pathology.[12]
In our case also, with these camouflaging clinicoradiological features, liver biopsy and thereby histopathological evaluation was done for confirmative diagnosis. The amyloid characteristically appears as acellular pale eosinophilic and amorphous material on routine hematoxylin and eosin stain. When stained with Congo red, it showed salmon red color on light microscopy and apple-green birefringence under polarized microscopy. Further typing of amyloid deposited can be done using ancillary histopathologic technique like immunohistochemical staining for monoclonal proteins such as kappa, lambda chains, and protein SAA.[13] In our case, the mystery behind the type of amyloid deposited was solved by and deceive our perception. Indian J Med Paediatr Oncol immunohistochemistry.
Hepatic amyloid deposits in systemic AA amyloidosis are typically associated with significant amyloid deposition in other organs when present carries a poor prognosis. The other organ involvements can present as nephrotic syndrome, congestive cardiac failure, high bilirubin, and high platelet count which are some of the predictors of poor prognosis in these patients.[14] Mean survival rate in patients with hepatic amyloidosis is 9 months.[15] Our patient also had the nonobstructive type of hypertrophic cardiomyopathy on 2D echo and succumbs to death within 6 months of the initial diagnosis of hepatic amyloidosis.


Hepatic amyloidosis can present as clinicoradiological dido and requires liver biopsy and ancillary histopathological techniques not only to attest the diagnosis but also for typing of amyloidosis. The presence of cardiac involvement is associated with poor outcomes in these patients.
Subscribe now for latest articles and news.